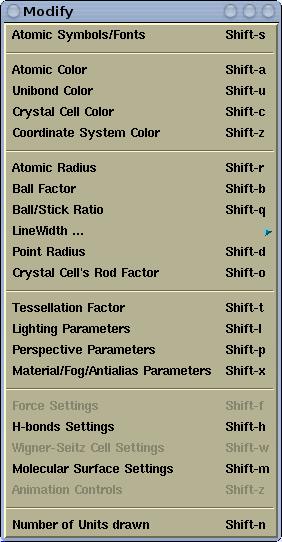
Various display parameters can be modified via the
Modify menu. Here is its
snapshot for
XCrySDen-v1.4:
Atomic synbols/fonts can be set and/or modified via menu:
Modify-->Atomic
Symbols/Fonts . There are two types of settings: (i)
global and (ii) custom. The global settings (font and color) apply
to all default atomic labels, i.e., those that were not customarily
set. There are two different font colors: (i) bright font color,
and (ii) dark font color. The first is used for display modes such
as ball-and-sticks and spacefills, where the labels are written on
the atomic-balls. The latter is used for display mode such was
lines (wire-frame). Here the atomic labels are put beside the
atomic positions.
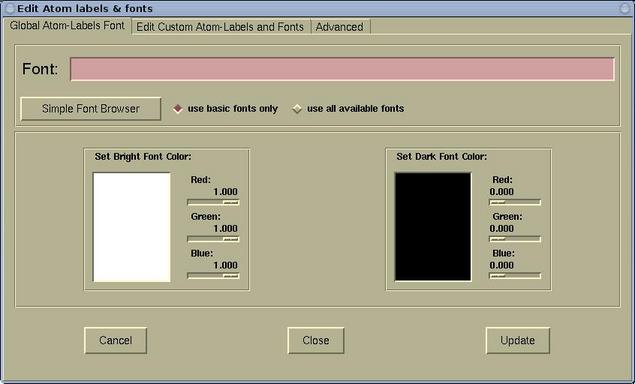
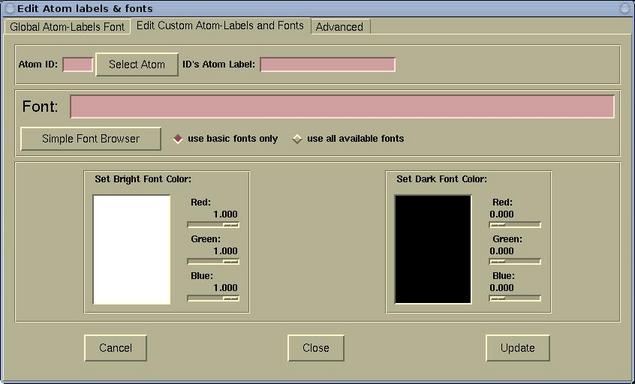
The Edit Atom labels & fonts window has three
pages:
- The Global Atom-Labels Font page is used for setting the
font and color of default (global) atomic labels. Here is this
page:
- The Edit Custom Atom-Labels and Fonts page is used for
setting the custom atomic labels. First a given atom must be
selected (via the [Select Atom] button). For the selected
atom, the label, font, and font-color can be set. The whole process
can then be repeated for other atom. The labels that were defined
in this was are called "custom atom-labels".
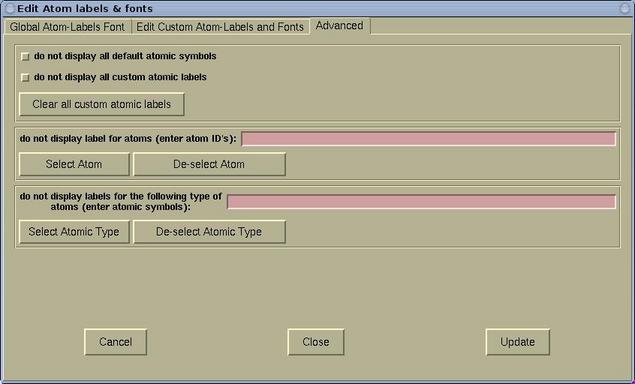
- The Advanced page is used for further label
manipulation. Here it is possible to toggle the display of default
and custom labels. It is possible to toggle the display of labels
for given individual atoms or for given atomic types.
An atomic color can be set via menu:
Modify-->Atomic Color
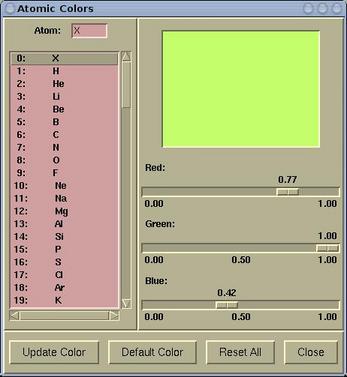
To modify an atomic color do the
following:
Select a particular element from the left listbox and set the color
by dragging the Red/Green/Blue scales. When the color suit your
needs, press the
[Update Color] button, otherwise the
setting will be lost. Buttons at the bottom of the
Atomic
Colors window have the following function:
| [Update Color] |
updates the atomic color of selected
element
|
| [Default Color] |
resets the atomic color of selected element
back to its default value
|
| [Reset All] |
resets all atomic colors back to default
values
|
| [Close] |
closes the Atomic Colors window,
otherwise does nothing
|
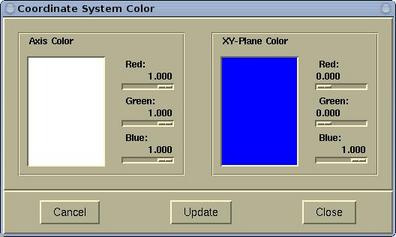
The color of the Coordinate system can be set via menu:
Modify-->Atomic
Radius .
Atomic radius and other related items can be set via:
Modify-->Atomic
Radius
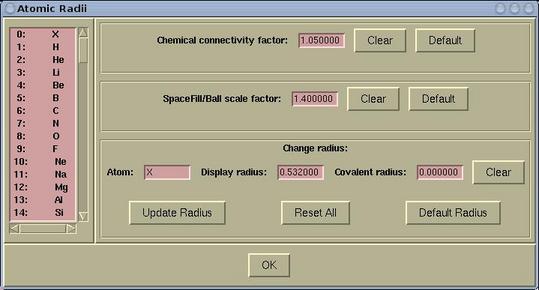
The following parameters can be set here:
|
chemical connectivity factor |
determines, whether to draw the bond between
two atoms. The bond between two atoms is drawn when the distance is
less than the sum of their covalent radii times the chemical
connectivity factor
|
| spacefill scale factor |
determines the size of the spacefills and
balls. The spacefill radius is:
rspacefill = spacefill_scale_factor *
ratomic
where ratomic is a display radius of the
corresponding atom.
|
| display radius |
it determines the display-size of a given
atom.
|
| covalent radius |
well, everybody knows what it is ...
|
To modify either chemical connectivity factor
or spacefill scale factor do the following:
In the corresponding entry widget enter a value and press the
[OK] button, which is at the bottom of the window.
To modify the display radius or the
covalent radius do the following:
First, select a particular element from the left listbox and enter
a new value in the corresponding entry. Then press the [Update
Radius] button, otherwise the setting will be lost.
Buttons in the Atomic Radii window have the following
function:
| [Clear] |
clears a value in the entry to null
string
|
| [Default] |
sets a default value
|
| [Update Radius] |
updates the covalent radius of selected
element
|
| [Reset All] |
resets the covalent radius of all elements
back to default values
|
| [Default Radius] |
resets the covalent radius of selected element
back to its default value
|
| [OK] |
updates chemical connectivity factor
and spacefill scale factor and closes the Atomic
Radii window
|
A tessellation factor can be set via menu:
Modify-->Tessellation Factor . Then one
simply enter a new value. Quite trivial.
Here I would like to explain what the tessellation
factor is. This factor determines the quality of the structure
display (balls, bonds, vectors). It has nothing to the with the
quality of an isosurface display. The greater the value of
tessellation factor the better the quality of display.
According to the value of tessellation factor the
quality of the display can be classified as:
| quality |
tessellation factor |
| bad |
1-20 |
| moderate |
20-40 |
| good |
>40 |
Please note that
tessellation factor is just a hint for
XCrySDen, since the
program will make some tuning according to the size of the
displayed structure (for big structures the tessellation factor is
internally reduced by some factor).
A setting of the force display can be edited via menu:
Modify-->Force
Settings . Forces are rendered as arrows and the
lengths of the display arrows represent the force magnitudes. Here
one can specify the (i) scale function, (ii) threshold, and (iii)
length factor. In addition, the display attributes of force arrows
ban be set (aspect and color).
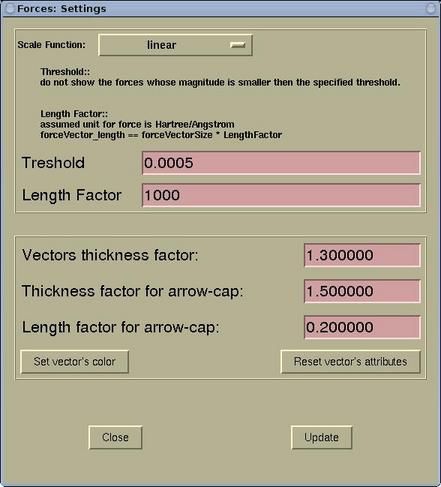
| Scale Function |
can be set to: linear, logarithmic, square
root, cubic root, exponential, and exp(x*x). According to my
experiences the most useful scale for the visualization of forces
is linear. However if one has a structure where the force sizes
range over the orders of magnitude, then the logarithmic scale
would be better. In this case a force of 0.01 Ryd/Bohr would be
displayed as a vector of 2 arbitrary length units in size, while
the force of 0.001 Ryd/Bohr as 1 arbitrary length units in
size.
|
| Threshold |
don't display forces below a given
threshold
|
| Length factor |
controls the visual length of the displayed
force-vectors. The assumed force unit is Hartree/ANGSTROM and a
force of 1 Hartree/ANGSTROM will be rendered as 1 ANGSTROM long if
the length factor is equal to 1. The actual displayed length can be
calculated as:
length = lengthFactor * forceMagnitude
|
| Vector thickness factor |
determines how thick is the force arrow.
|
|
Thickness factor for arrow-cap |
determines how thicker is the arrow-cap with
respect to arrow's line.
|
| Length factor for arrow-cap |
determines the length of the arrow-cap in
terms of the fraction of the arrow's length.
|
| [Set vector's color] |
Here the color for the force arrows can be
set.
|
Warning: press the [Update] button to load the new
setting and to update the display.
A setting of the display of the hydrogen bonds can be edited via
menu:
Modify-->H-bonds
Settings . The display of the H-bonds can be
controlled by the following parameters in the
H-bonds:
Settings window.
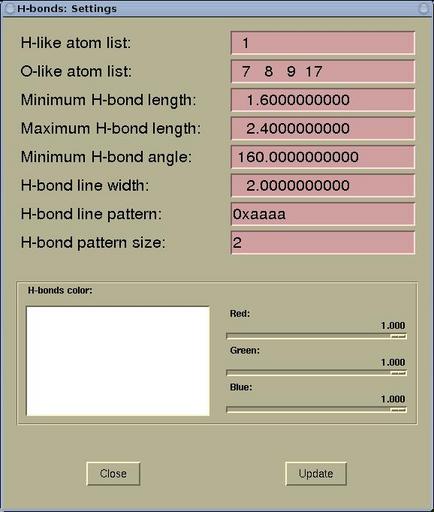
| H-like atom list |
here the atomic numbers for the H-like atoms
are specified. In fact, it should be only hydrogen, but having the
possibility to enter also other atoms make larger display
flexibility. ( For example, there can be two different
types of H atoms in a given molecule, and one would like to display
them differently. Hence in the structure file (such as XSF) the two types can
be represented by H and He.) |
| O-like atoms list |
the list of the electro-negative atoms (enter
atomic numbers) such as O, N, F that can form the hydrogen
bond
|
| Minimum H-bond length |
is a minimal allows length of the H-bond (used
to distinguish between the normal chemical bond and the hydrogen
bond)
|
| Maximum H-bond length |
the maximum length between the H and the other
atom to be still considered as the H-bond
|
| Minimum H-bond angle |
the minimum A-H---B bond angle to be still
considered as the H-bond
|
| H-bond line width |
the thickness of the displayed H-bond (the
H-bond is displayed as line)
|
| H-bond line pattern |
the dashing pattern of the lines that
represent the H-bonds (in hexadecimal form, e.g.
0xeafa)
|
| H-bond pattern size |
the dash length of the lines that represent
the H-bonds
|
| H-bonds color |
the display color of the H-bonds
|
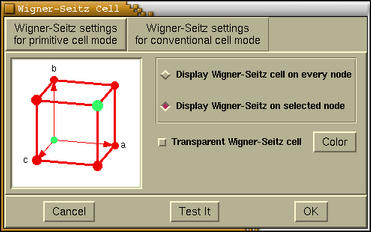
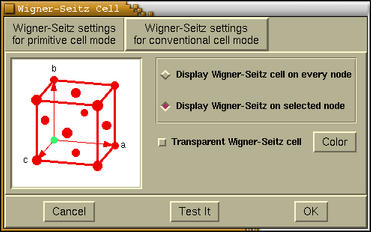
A setting of the Wigner-Seitz cell display can be edited via menu:
Modify-->Wigner-Seitz Cell
Settings . The following window pops-up when this
options is selected.
At the top of this window two tab buttons are located. These are
[Wigner-Seitz setting for primitive cell mode]> and
[Wigner-Seitz setting for conventional cell mode]. Under the
each tab we can set the display of the Wigner-Seitz cells for the
appropriate unit-cell display-mode.
Below the tab buttons a frame with several widgets is located.
On the left the appropriate lattice-type is displayed, while on the
right the following items widgets (widgets) are mapped:
- [Display Wigner-Seitz cell on every node]
- displays the Wigner-Seitz cell on very lattice point.
- [Display Wigner-Seitz cell on selected node]
- displays the Wigner-Seitz cell only on selected lattice points.
This selection is done by mouse-clicking the lattice points
displayed on the left side unit-cell-display window.
- [Transparent Wigner-Seitz cell]
- toggles the transparency of the displayed Wigner-Seitz
cells.
- [Color]
- sets the color of the Wigner-Seitz cells.
Warning:
The [Test It] button should be pressed to load new setting
and update the display of the Wigner-Seitz cells. The
[Cancel] button disables the display of Wigner-Seitz cells,
while [OK] button merely closes the Wigner-Seitz
Cell window (Test-It button should be pressed prior
to load the setting).
A setting of the Molecular Surface Settings can be edited via menu:
Modify-->Molecular Surface
Settings .
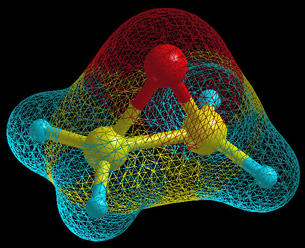
In
XCrySDen this term is
used for a special kind of plots. One purpose of this plots is yet
another display-mode of a molecule. Unlike the ball&stick it is
not build from simple graphical primitives like spheres and
cylinders, but rather resembles the molecular charge density
display. An oxirane molecule is displayed on the below figure in
the ball&stick display-model with the molecular surface, which
is displayed in
wire mode.
The molecular surface is build with the following recipe:
- on each atomic center put the following
Gaussian function:
fa(x) = 2 * exp[
-ln(2)/ra2 * |xa -
x|2 ],
where the x_a is the position of the atom A,
and r_a is some specific radius for the atom A
(usually covalent or van der Waals radius). This Gaussian function
has the following properties: (i) at the nucleus center A its value
is 2, and (ii) at the |x_a - x| = r_a it
equals to 1 (i.e. 50% of its maximum value).
- calculate the 3D grid of points inside an appropriate box,
which embeds the molecule.
- specify an isovalue for the molecular surface, which should be
in the [0,2] range (a good value is 1).
- triangulates and render the isosurface.
The following window appears when the
Modify-->Molecular Surface Settings
menu option is selected.
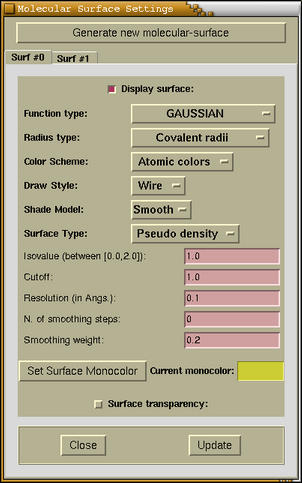
At the top of the window the
[Generate new
molecular-surface] button is located. It generates new
molecular surface. The properties of each newly generated molecular
surface are set to some preset-default values, but you can control
them by pressing an appropriate tab button. The tab buttons are
located just below the
generate button. There are plenty
of widgets on each tab-page.
- [Checkbutton: Display surface]
- Toggles the display of current molecular surface.
- [Optionmenu: Function type]
- Here we can chose among different functions. These are the
functions alike the above Gaussian
function. The following functions beside the above Gaussian are
available:
- EXPONENTIAL
- an exponential function, with the similar properties as above
Gaussian function, namely: (i) at the nucleus center A its values
is 2, and (ii) at the
|x_a - x| = r_a it
equals to 1 (i.e. 50% of its maximum value).
- constant GAUSSIAN
- alike GAUSSIAN function, but the
r_a for all the
atoms is taken to be constant and is set to 1 ANGSTROM.
- constant EXPONENTIAL
- alike EXPONENTIAL function, but the
r_a for all
the atoms is taken to be constant and is set to 1 ANGSTROM.
- distance FUNCTION
- in each point of the 3D grid the distance to the nearest atom
is stored. This is useful for the gap
analysis.
- [Optionmenu: Radius type]
- Chooses the radius-type of
r_a among covalent and
van der Waals radius
- [Optionmenu: Color Scheme]
- Chooses among Atomic colors and Monocolor.
When the surface is colored in atomic-colors then it inherits the
color from the nearest atom. On the other hand the mono-colored
surface has a single color, which can be set by pressing the
[Set Surface Monocolor] button.
- [Optionmenu: Draw Style]
- Sets the draw-style of the surface (solid, wire, dot).
- [Optionmenu: Shade Model]
- Sets the shade-model of surface (smooth, flat).
- [Optionmenu: Surface
Type]
- Sets the surface type, which can be either a
Pseuodo-Density or Gap analysis. The
Pseuodo-Density is the type of surface that was explained
above. It name comes from the fact that
these plots look alike the charge-density plots. On the other hand,
the Gap analysis is available only for periodic
structures. These plots are useful for the examination of open
structures as they really plot the holes (i.e. vacancies) inside
the structure. A distance Function function is useful for
this purpose, but other functions will do as well. Here is an
example of a Chabazite crystal:
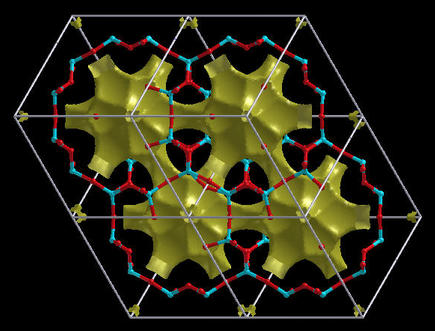
Note: for Gap analysis the isovalue is not confined
to the range [0,2].
- [Entries: Isovalue, Cutoff, Resolution, N. of smoothing
steps, Smoothing weight]
- The isovalue is the value at which the molecular
surface will be tessellated; the cutoff is the margin
around the bounding-box that embeds the molecule; the
resolution specify the resolution for the grid calculation
- the quality of molecular surface depends heavily on this
parameter. The last two parameters are for surface-smoothing. This
doesn't work well, but maybe for some cases should produce a nicer
surface.
- [Button: Set Surface Monocolor]
- Sets the color of the molecular surface for the
monocolor scheme.
- [Checkbutton: Surface transparency]
- Toggles the transparency of the surface
- [Button: Close]
- Closes the Molecular Surface Settings window.
- [Button: Update]
- Important: this button should be pressed to load new
setting and update the surface display
The animation can be controlled by the
Animation Control
Center window, which is accessible via
Modify-->Animation Settings .
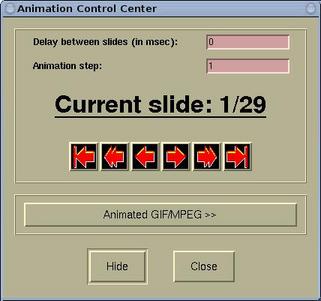
On this window we can set the delay (in msec) between slides, the
animation step, and there are the playing buttons (from left to
right:
to the first, animate backward, one step back, one step
forward, animate forward, and
to the last).
To enhance the performance of the animation we can either switch
to Lighting-Off mode or reduce the tessellation factor.
The [Hide] button hides the Animation Control
Center window. Hiding means that window is iconified, i.e. the
window disappears and its icon appears on XCrySDen main render window. By
mouse-cliking the icon the window will appear again.
The [Animated GIF/MPEG >>] button is enabled only
when the GIF and/or MPEG encoders are defined in the the $HOME/.xcrysden/custom-definitions file. By
pressing this button, new information will appear on the window
aimed at creating animated-GIF and MPEG movies. Read More ...
The number of displayed crystal unit cells can be set via
Modify-->Number of Units
Drawn menu, where we simply specify the number of
unit cells to be drawn in each crystallographic direction.
BEWARE: don't specify to large numbers, for example
10x10x10, since it will either take a long time to render the
crystal or the program will run out of memory.

![[Figure]](img/xcrysden-picture-small-new.jpg)